

Summary:
Discover how medical device manufacturers can build a strong, compliant CAPA process using digital QMS tools. Learn common CAPA pitfalls, real-world examples, and practical ways to improve product quality, patient safety, and regulatory readiness through automation and risk-based management.
In the medical device industry, Corrective and Preventive Action (CAPA) is a structured process companies use to identify, fix, and prevent quality issues. It is what keeps medical devices safe, compliant, and trusted in the market.
But in an industry where precision and compliance define success, a weak or manual CAPA can quietly expose companies to serious regulatory risks.
Yet, many medical device manufacturers still view CAPA as a reactive formality rather than a proactive driver of product quality and operational excellence.
Between 2013 and 2019, 72% of medical device warning letters cited failures to “establish and maintain” a proper CAPA process. That implies most compliance breakdowns are not just about poor products; they’re about broken systems.
And one thing is for sure: CAPA is not just for compliance; it impacts your product quality, patient safety, decision-making, and even business stability.
So, why is it more important than ever to strengthen your CAPA process?
Let’s explore what CAPA in the medical device industry truly means—and how you can streamline or digitize it with a robust QMS.
See how a robust QMS simplifies your quality process
Schedule DemoWhat is CAPA in the Medical Device Industry?
Corrective and preventive action is a structured process that identifies, resolves, and prevents quality issues that can impact the quality of medical devices. The objective of CAPA is to eliminate the immediate problem and implement adequate measures to prevent its recurrence.
The medical device industry operates in a highly regulated environment. Medical devices interact directly or indirectly with human bodies, where even a minor defect can have life-threatening consequences for patients.
Quality issues, such as design verification failures, repeated supplier nonconformances, out-of-spec raw materials, equipment calibration and maintenance failures, or incorrect testing methods, can impact patient safety and potentially lead to reputational damage.
A good CAPA makes sure that if there is a quality problem, the company not only fixes it but also looks at data to find patterns and the root cause, plans and executes suitable corrective and preventive actions, and keeps a record of the whole process for tracking and compliance.
Regulatory Compliances That Mandate CAPA for Medical Device Industries
Regulatory bodies worldwide require medical device manufacturers to plan and implement a robust CAPA process, ensuring that issues are detected, investigated, corrected, and prevented through a documented and traceable method. The following key compliances clearly define CAPA expectations.
1. ISO 13485:2016
ISO 13485:2016 is an international standard for quality management to regulate the medical device industry.
It requires organizations to establish a documented process to identify nonconformity, investigate the cause, and take corrective and preventive actions. This ensures that every issue is systematically addressed by the quality team, and its effectiveness is verified.
2. FDA 21 CFR Part 820
FDA’s Quality System Regulation (QSR) sets good manufacturing practice (cGMP) requirements for all medical device manufacturers.
FDA 21 CFR Part 820 mandates CAPA under Subpart J, which talks about corrective and preventive actions and instructs manufacturers to analyze data, investigate the actual cause of the nonconformities, and take actions to prevent similar incidents from occurring in the future.
3. EU MDR 2017/745
The European Union Medical Regulation (EU MDR) establishes a detailed framework that regulates the medical device industry.
It emphasizes CAPA within its Post-Market Surveillance (Article 83-86) requirements. Unlike other compliances, it focuses more on monitoring product performance, identifying trends, and implementing CAPA based on real-world feedback.
4. ISO 14971
ISO 14971 serves as a critical guideline for medical device manufacturers. Primarily, it regulates risk management that directly links to CAPA by requiring manufacturers to control and monitor risks throughout the product lifecycle.
Why CAPA Fails in Many Medical Device Companies?
Even with a well-defined procedure, many medical device manufacturers struggle to make their CAPA truly effective. However, the problem lies not in the process itself, but in its execution.
When CAPA becomes a reactive exercise rather than a proactive quality tool, it fails to deliver meaningful or lasting improvement. Here are some of the most common reasons why CAPA initiatives fall short in the medical device industry:
Inadequate Root Cause Analysis:
Often, teams focus on symptoms rather than the underlying causes. Without a detailed root cause analysis, actions only provide temporary fixes, allowing the same issues to reappear later.
Overcomplicated or Manual Processes:
Paper-based or disconnected processes slow down investigations, create data silos, and make tracking even more difficult. This leads to delays, incomplete documentation, and missed follow-ups.
Lack of Risk-Based Prioritization:
When every issue is treated with the same level of urgency, it affects resource allocation and prevents teams from focusing on the problems that most impact patient safety and compliance.
Weak Verification of Effectiveness:
Many CAPAs are closed without confirming whether the corrective actions truly prevented recurrence. And without measurable effectiveness checks, it becomes difficult for organizations to validate real improvement.
Poor Integration with Other Quality Processes:
CAPA cannot succeed in isolation. Without links to audits, nonconformances, inspections, etc., it operates in silos, limiting visibility, traceability, and overall effectiveness.

Get practical insights and expert tips to strengthen your quality system.
Download NowWhat Triggers CAPA? Common Examples from Medical Device Manufacturing
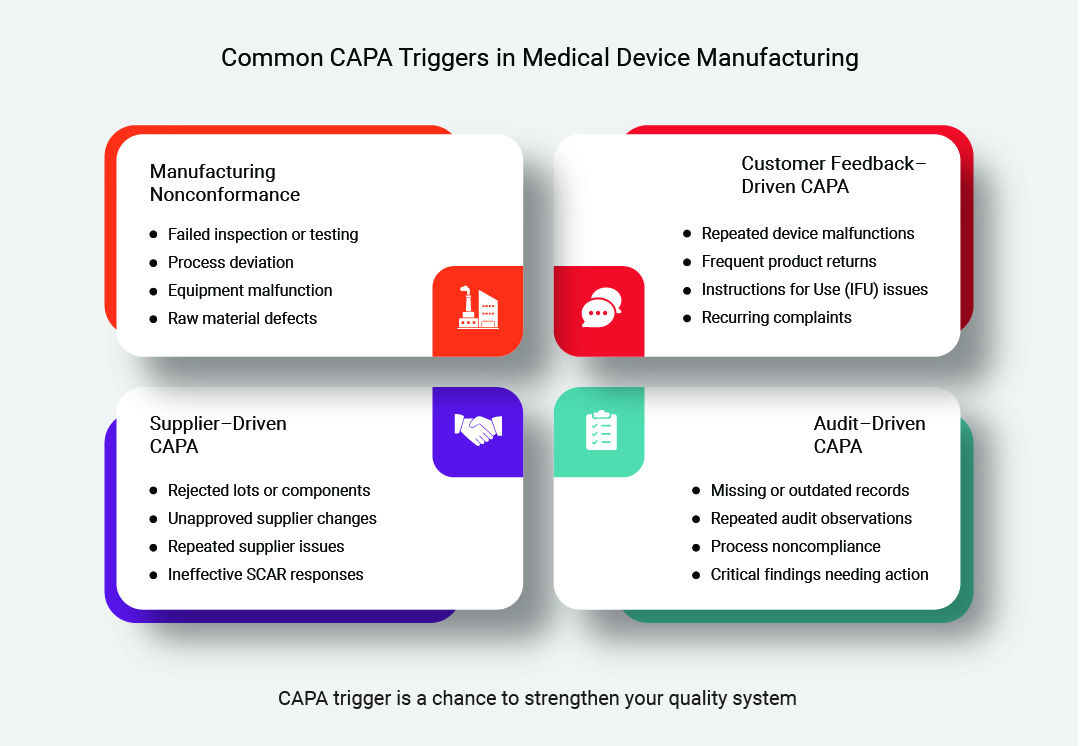
CAPA Triggered by Manufacturing Nonconformance
Manufacturing nonconformances occur when a product, process, or even raw material in the production environment fails to meet predefined standards required to achieve quality benchmarks.
The primary reasons might involve a deviation in any of the following: product characteristics, process parameters, equipment performance, etc. Let’s explore some examples of manufacturing nonconformance.
- If the physical product fails inspection or testing
- If a process deviates from a pre-approved or validated process
- If a machine or equipment used in production does not function correctly
- Raw materials or components used in production fail to meet the set criteria
Customer Feedback–Driven CAPA
Customer feedback-driven CAPAs are initiated when the after-sales market data or customers’ responses highlight recurring problems or potential risks faced by the customers while using medical devices.
The source of such issues can be your complaint record, service reports, returned products, or trend analysis conducted by the quality team. A few examples that may trigger a CAPA can be:
- Customers repeatedly report the same type of device malfunction or failure in the field
- Product returns and replacements show a consistent failure mode or pattern
- Feedback indicates user confusion or difficulty interpreting instructions for use (IFU)
- Complaint trends highlight an increasing rate of similar issues
Audit–Driven CAPA
Audit-driven CAPAs are triggered when internal, external, or regulatory audits find gaps in compliance, documentation, or process execution. These findings point to systemic weaknesses in the Quality Management System (QMS) that need formal investigation and long-term corrective measures. Some common examples may include:
- Auditors identify repeated instances of incomplete or missing quality records
- Procedures or work instructions are found to be outdated
- Internal audits highlight the same observations across multiple audit cycles
- When external audits identify a critical nonconformity requiring documented follow-up actions
Supplier–Driven CAPA
Supplier-driven CAPA is needed when quality issues related to purchasing raw materials or components affect the quality of a medical device. Such CAPA often focuses on supplier process controls, material consistency, and compliance with agreed quality requirements. Here are some examples that can trigger supplier-related CAPA:
- Multiple lots of a supplied component fail incoming inspection due to the same defect
- Supplier changes a manufacturing process or material without proper notification
- Recurring quality issues are found during supplier audits or performance reviews
- Supplier Corrective Action Request (SCAR) reveals improper RCA or ineffective corrective measures
Building a Robust CAPA Process that Fits Best for Medical Device Manufacturers
A standard CAPA process is followed across most industries, covering steps from issue identification to documentation and trend analysis.
However, how can CAPA be strengthened in the medical device industry, where it serves as a business-critical pillar of quality?
According to the Medical Device Innovation Consortium (MDIC), manufacturers who adopted a risk-based CAPA model reduced their improvement-implementation time by up to 80%, demonstrating that tailoring CAPA depth to risk can dramatically boost efficiency without compromising compliance.
Here’s how medical device manufacturers can build a robust, risk-based CAPA framework—and how a digital QMS can make it faster and more traceable:
1. Issue Identification and Reporting
Timely detection and transparent reporting ensure that potential risks are identified early, thoroughly investigated, and resolved before they escalate. Ensure all nonconformances, complaints, and audit findings are captured systematically and communicated with quality teams.
How a digital QMS supports this:
- Centralized issue logging with automated form templates for consistent data capture
- Integration with audit and nonconformance modules to ensure traceability
- Automated notifications to quality teams for prompt action
- Dedicated dashboards for monitoring issue trends across manufacturing and post-market stages
2. Risk Evaluation and Prioritization
Every reported issue must be assessed for potential impact on patient safety, product quality, and regulatory compliance. However, not all issues carry the same level of risk, so prioritizing them can help with resource allocation and focusing on high-risk issues at the earliest opportunity.
In practice, with a digital QMS, you can:
- Use built-in risk scoring matrices to quantify severity
- Automatically link issues to relevant risk management files and design history records (DHRs)
- Assign risk-based priorities for CAPA initiation and escalation
3. Initial CAPA Planning
Initial CAPA planning defines how the team will address a detected issue. Once an issue is prioritized, a structured CAPA plan should be made to outline the scope, responsibilities, objectives, and timelines.
Using a digital QMS, you can:
- Create CAPA workflow templates with assigned roles and approval hierarchies
- Attach supporting documents, test results, or audit data directly to the CAPA record
- Set automated reminders and escalation paths for overdue tasks
- Enable stakeholder review and sign-off through secure digital approval
4. Root Cause Investigation and Analysis
Effective CAPA relies on accurate root cause analysis (RCA) to prevent recurrence. RCA helps identify the true cause of the issue, not just its symptoms.
Digital QMS advantages:
- Provide structured investigation tools (5 Whys, Fishbone, or Pareto analysis) within the platform
- Centralize data from production, complaints, and audits for cross-functional analysis
- Maintain traceability between cause, effect, and evidence of documentation
- Facilitate collaboration among quality, design, and operations teams in real time
5. Execution of CAPA Action Plan
Actions are implemented according to approved timelines and must be monitored for progress and completion.
Digital QMS support includes:
- Real-time progress tracking with completion status indicators
- Automatic task assignment and escalation for overdue actions
- Integration with document control to update SOPs, work instructions, or batch records
- Comprehensive audit trails to demonstrate regulatory compliance
6. Verification and Validation of CAPA Effectiveness
Verification ensures that the CAPA is implemented, while validation confirms that it effectively resolves the root cause without introducing new risks.
With digital tools, you can:
- Capture evidence of CAPA effectiveness (test results, inspection data, trend reports)
- Define and track key metrics, such as defect rates or complaint frequency
- Automate validation reviews with digital signatures and timestamped records
7. Documentation, Review, and Closure
Comprehensive documentation ensures traceability and regulatory readiness. Every CAPA must undergo QA review before closure.
Digital QMS benefits:
- Central repository for CAPA history, evidence, and approval records
- Automatic generation of closure reports
- Version control to manage document updates aligned with CAPA outcomes
- Seamless audit-readiness with searchable, traceable records
8. Trend Analysis and Ongoing Improvement
Trend analysis identifies recurring patterns and systemic gaps, feeding insights back into continuous improvement initiatives.
Through a digital QMS, you can:
- Analyze CAPA data across product lines, processes, or suppliers
- Generate automated dashboards highlighting recurring root causes
- Link trend insights to quality planning activities
- Foster a proactive quality culture by converting insights into strategies
How Can BizPortals QCFlow Be a Game Changer?
A well-executed CAPA system is a foundation for trust, safety, and continuous improvement in medical device manufacturing. However, leveraging a digital QMS, manufacturers can turn CAPA into a true driver of quality.
BizPortals QCFlow, a secure QMS software powered by Microsoft 365, can empower this transformation. It simplifies CAPA management through automation, integration, and real-time visibility.
Its configurable workflows, risk-based tracking, and connected modules for different quality processes help medical device manufacturers strengthen compliance, improve traceability, and accelerate corrective actions—all within a secure, user-friendly Microsoft 365 environment.
See the difference for yourself. You can connect with our experts and experience a live demo of BizPortals QCFLow to explore how it can simplify and strengthen your CAPA process.
Experience BizPortals QCFlow in action—tailored for you.
Book a DemoFAQs
-
1. What is CAPA in medical device manufacturing?
CAPA (Corrective and Preventive Action) is a process used to identify, resolve, and prevent product or process issues that could affect medical device safety or compliance.
-
2. Why is CAPA important for medical device manufacturers?
CAPA ensures product quality, patient safety, and regulatory compliance by systematically addressing nonconformances, reducing risks, and preventing recurrence of issues in medical devices.
-
3. What are the main stages of the CAPA process?
The CAPA process includes issue identification, risk evaluation, root cause analysis, planning, implementation, effectiveness verification, documentation, and ongoing improvement tracking.
-
4. What are common challenges in CAPA implementation in the medical device industry?
Common challenges include poor root cause analysis, incomplete documentation, delayed response, lack of follow-up, and insufficient verification of CAPA effectiveness.
-
5. What documentation is required for CAPA closure in the medical device industry?
Required records include investigation details, root cause evidence, corrective and preventive actions, verification results, approvals, and closure confirmation to demonstrate complete and effective CAPA implementation.
-
6. How often should CAPA effectiveness be reviewed?
CAPA effectiveness should be reviewed after implementation, during follow-up audits, and periodically in management reviews to confirm long-term issue resolution.

